
[youtube]https://www.youtube.com/watch?v=fcUzFlk4VKg[/youtube]Ethnic minorities, low-income individuals and those who live in rural areas may be missing out on early diagnosis and state-of-the-art treatments for cystic fibrosis, according to Emory University researchers. That’s a serious problem because such measures can both lengthen and improve life.
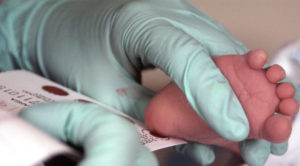
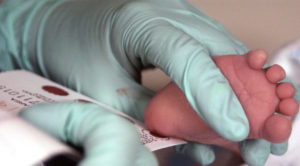
Heel-stick tests for newborns have been around for more than 50 years, and in 2007 Georgia added cystic fibrosis (CF) to the list of more than 30 serious health conditions detected by this method.
The test, however, isn’t perfect. It picks up 23 different abnormal changes in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, many of which can cause serious problems in multiple organ systems, but all of these mutations were identified by studying Caucasian people. So the test may miss genetic changes that lead to CF in patients of other ethnicities.
The ethnic imbalance in research is not totally arbitrary. Ninety-four percent of cystic fibrosis cases diagnosed in the United States are in Caucasians.

“Cystic fibrosis is predominantly a disease of patients of European ancestry,” said pulmonary specialist Dr. Seth Walker, assistant professor of medicine at Emory University and director of the Emory Adult Cystic Fibrosis Program. “The incidence is much smaller in blacks and Hispanics and Asians, but it’s still there. When we find it in these populations, they often have different mutations.”
There’s still more we need to learn about CF and ethnicity. New findings in the January 2016 issue of the Journal of Molecular Diagnostics from Stanford University show that non-Caucasian cystic fibrosis patients have CFTR mutations not included in the newborn screening test. Because of this, Walker suspects there is a disparity in diagnosing cystic fibrosis among ethnic minorities, but he can’t be sure due to lack of data on CF in these populations.
“We don’t have any clear data on how many are being missed, because we don’t know how many people are out there,” said Walker. “But certainly it’s much less common for anyone to think of the diagnosis in the non-white population. Unfortunately, we still find some doctors out there where patients have brought [the diagnosis] up themselves and are told, ‘You can’t have CF, you’re black.’ ”
Over time, the disease causes a buildup of thick, sticky mucus in the lungs, making breathing difficult. It also interferes with pancreatic function, leading to digestive difficulties. Proper medical treatment slows this process dramatically. In the United States, cystic fibrosis is the second most common inherited disorder that begins in childhood and shortens life expectancy, according to the CDC. (Only sickle cell anemia strikes more often.)
Major treatment centers
Of the approximately 1,000 Americans diagnosed with cystic fibrosis each year, about 30 reside in Georgia, according to Children’s Healthcare of Atlanta. Better medications and management techniques have doubled the average lifespan of CF patients from age 18 to age 40, Walker said, but only for patients fortunate enough to be treated aggressively.
Although non-white babies are most likely to be missed by newborn screening tests, location and income level are also powerful determinants of how well they respond to treatment.
Walker said the best place for care is a specialized center accredited by the Cystic Fibrosis Foundation, a national nonprofit organization focused on cystic fibrosis research and quality of care for CF patients. Georgia has CFF-accredited programs at Emory University, Children’s Healthcare of Atlanta and Augusta University, and adult programs at Emory and in Augusta. But there’s a problem in getting low-income patients and patients who live far away to these clinics for treatment.
Although medical treatment is covered by Georgia’s Medicaid system, low-income patients — even those who live in Atlanta or Augusta — may have trouble getting the transportation they need for frequent clinic visits.
Medicaid does provide state-funded transport to and from treatment facilities, but only if the patient has no other means of transportation or when an accessible means of transport would harm the patient, according to the state’s Department of Community Health.
Some patients may travel many hours to see cystic fibrosis specialists, and even go out of state. However, patients with government-sponsored insurance, including Georgia Medicaid, PeachCare for children or Tricare for military families, must be treated at a Georgia facility, even if a CFF-accredited center in another state is closer to their home. As a consequence, “oftentimes these patients go without,” Walker said.
Even when low-income patients are prescribed the same medicines as their wealthier counterparts, they may not respond as well to treatment.
“It doesn’t seem to be a difference in the medications they’re receiving,” said Walker; “they’re being prescribed the same medications everyone else is. So this may simply be a factor of poverty: that being poor has worse outcomes for your health and mortality.”
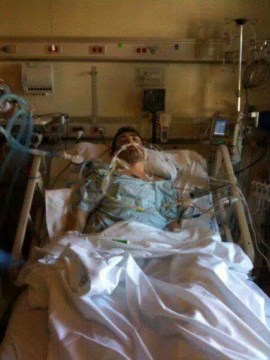
Joseph McCanne, 37, of Henry County is fortunate. Not only does he live close to Atlanta’s cystic fibrosis clinics, he also felt a renewed quality of life following a double-lung transplant at age 31.
“Up until 2009, right before I had a lung transplant, I was going to the hospital about every month [to] month-and-a-half for two weeks at a time,” said McCanne. “My lung function had dropped to 13 percent.”

Six years after his transplant, McCanne said cystic fibrosis doesn’t limit him at all. He still takes anti-rejection drugs for his lungs and pancreatic enzymes to help digest meals, but “it’s a world of difference,” he said. “Being able to do anything everybody else can do, I really feel like I don’t have a disease.”
Walker believes the continued inclusion of cystic fibrosis in newborn screening tests will mean better long-term outcomes.
Data from Colorado and Wisconsin — states that have tested newborns for cystic fibrosis since 1987 and 1994, respectively — have shown that patients treated for the disease before they show symptoms have better outcomes than patients who seek treatment months or years after symptoms begin. Implementing the findings of Stanford’s researchers and others who study cystic fibrosis in minority and rural populations could increase these positive outcomes.
“We’re finding people before they have disease,” Walker said. “They can start aggressive therapy before they have daily symptoms,” leading to extended lifespan and better quality of life.
Sandra L. McGill is a second-year master’s student in Health and Medical Journalism at the University of Georgia’s Grady College of Journalism and Mass Communication in Athens.